

Systemic Lupus Erythematosus (SLE)
Last updated: October 28, 2014
Synonyms: Lupus, lupus erythematosus, SLE
ICD-9 Codes: SLE 710.0; discoid, 695.4; lupus anticoagulant 286.5; drug-induced lupus 695.4; lupus nephritis 583.81.
ICD-10 Codes: SLE M32; discoid L93.0; subacute cutaneous lupus L91.0; lupus anticoagulant syndrome D68.62; antiphospholipid syndrome D68.61; drug-induced lupus, M23.0; lupus nephritis M32.14
Definition: SLE is a systemic autoimmune disease characterized by autoantibody production, inflammation, and tissue damage that can affect various organ systems. There is a large spectrum of disease activity and severity: some patients with SLE have relatively mild disease, while others experience severe morbidity and accelerated mortality. The characteristic laboratory finding of SLE is the presence of autoantibodies that react with various components of the cell nucleus; antinuclear antibodies (ANA). The presence of multiple autoantibodies is one of the defining characteristics of SLE, although the pathophysiologic role of some autoantibodies remains unknown. Some end-organ involvement in SLE involves autoantibody driven deposition of immune complexes. The presence of specific autoantibodies has some correlation with end organ involvement and prognosis.
Etiology: The etiology is unknown. Given the significant female preponderance of SLE, sex steroids are presumed to play a key role in disease expression. Genetics play some role; monozygotic twins are concordant for SLE in approximately 25% of cases, whereas dizygotic twins and other siblings are concordant in 5%. This implies that other environmental risk factor(s) are superimposed on a susceptible genetic background. Certain major histocompatibility complex alleles (e.g., HLA- B8, DR2, DR3) are associated with a slightly greater risk of developing SLE. In addition, some patients with SLE have null alleles for complement protein C4. Although this may not be reflected in low serum C4 concentrations, it may affect the patient’s ability to remove immune complexes effectively. A very strong, albeit rare (<100 cases reported) genetic predisposition to SLE is complement protein C1q deficiency, where almost all patients develop SLE. In addition, autoantibodies against C1q are commonly observed in SLE patients. Other genetic factors affect manifestations of disease. For example, allelic differences in cell surface receptors for the Fc portion of IgG have been shown to correlate with some end-organ involvement in SLE. There has been interest in the role of the type I interferon (IFN) system in SLE. Elevated levels and activity of INF-α have been shown to correlate with disease activity in SLE patients. This is sometimes quantified as the “interferon signature”, with increasing expression of a variety of proteins whose synthesis is potentially driven by IFN. Recently, there has also been interest a potential role in SLE for neutrophil extraceullar traps (NET; chromatin and enzymes released by extracellularly by neutrophils in response to infection). Increased NET formation and decreased processing of NETs have been observed in SLE. In common, immunologic alterations that predispose to SLE may interfere with the normal process of clearance of products from dying cells, that may then predispose to autoimmunity to normal constituents.
Demographics: Peak incidence of SLE is between 15 and 40 years of age. In this age group, women are affected approximately 10 times as commonly as men. This female predominance decreases among older patients. There is a racial disparity: patients of African descent have both a greater incidence of SLE and a tendency toward more severe disease. Asian and Hispanic patients also have been reported to have more severe disease than Caucasians. The overall population prevalence of SLE is approximately 25 to 50 per 100,000. Among some high-risk populations (e.g., young black women), the prevalence may be as high as four per 1,000.
Cardinal Findings: Characteristic clinical findings of SLE are shown in Table 1. The frequencies of end-organ involvement between populations and among patients with SLE differ substantially. This is relevant to the treatment of SLE, which is often guided by the particular constellation of clinical characteristics and the most severe end-organ involvement for a given patient. Some manifestations of SLE vary with race; for example, discoid skin lesions are more common and photosensitivity is less common among patients with SLE of African descent as compared to Caucasians. Some manifestations may be more common among patients with particular autoantibodies (e.g., renal disease in patients with anti-double stranded DNA antibodies). A number of patients with SLE have an overlap of signs and symptoms of other connective tissue diseases. Manifestations typical of scleroderma (sclerodactyly, interstitial pulmonary disease, digital vasculitis) and inflammatory myositis are not infrequently encountered among patients with SLE.
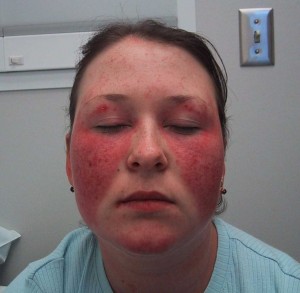
—Skin: Before the widespread availability of immunologic laboratory tests for SLE, dermatologic manifestations were perhaps the most characteristic finding of SLE. Indeed, before the early 1900s, SLE was considered a largely dermatologic disease, with internal organ involvement noted only on necropsy. Lupus (Latin for ‘wolf’) erythematosus refers to the red appearance of the inflammatory malar rash which was likened to a wolf bite. The malar rash, also known as the butterfly rash, is typically a maculopapular rash over the malar bones of the cheeks. The rash tends to spare the nasolabial folds, in contrast to seborrheic dermatitis which frequently crosses the nasolabial folds and is found at the forehead and eyebrow hairline. It can be difficult to differentiate clinically from some other facial rashes, including rosacea and polymorphous light eruptions. Not infrequently patients with dermatomyositis will manifest a malar rash. Histopathologically, biopsy of a lupus rash reveals granular deposition of immune complexes and complement in a band-like pattern at the dermoepidermal junction (the so-called lupus band test). In acute lupus, rashes such as the malar rash, clinically uninvolved skin also shows such deposits. In contrast, in discoid lupus skin lesions, immune deposits are only seen in involved skin. Unlike the malar rash and other dermatologic manifestations, discoid lupus may occur in the absence of systemic involvement. Another distinguishing feature of discoid lupus is that it tends to involve the supporting skin structures, such as hair follicles, and causes follicular plugging and alopecia. When discoid lesions resolve, they often result in residual scarring, altered pigmentation (e.g. vitiligo) and alopecia that can be disfiguring. By contrast, acute lupus lesions or subacute cutaneous lupus erythematosus (SCLE) lesions can resolve without scarring or other sequelae. Subacute cutaneous lupus erythematosus refers to annular or papulosquamous lesions associated with antibodies to Ro (SS-A) and usually occurs in sun-exposed areas of the arms and trunk. Most lupus rashes tend to be exacerbated by sun exposure, and intense sun exposure may also precipitate a flare of systemic disease in SLE patients. The treatment of lupus rashes depends on their severity and extent. For some patients, topical corticosteroid preparations may effectively control the lesions. Antimalarial medications, particularly hydroxychloroquine, can be effective for skin lesions.
—Renal: Kidney involvement is very common in SLE and may be associated with substantial morbidity and mortality. Almost all patients with SLE will be found to have some deposits of immune complexes and complement in the renal mesangium (WHO class II). However, such lesions may not precipitate renal inflammation and damage, and therefore may not require specific intervention. However, these early or mild lesions can progress to more serious lesions. Proliferative glomerular lesions (WHO class III or IV), which may be characterized by subepithelial, subendothelial, and intramembranous immune complex deposits and diffuse glomerular, as well as tubulointerstitial, inflammation, are of particular importance. Untreated, they often progress and cause renal failure. Purely membranous glomerulonephritis (WHO class V) that can occur and typically manifest with proteinuria; membranous lesions may also occur in combination with proliferative lesions. Patients with SLE with high titers of antibodies to double-stranded DNA (anti-dsDNA antibodies) are at greater risk of developing proliferative lupus nephritis, although the presence of such antibodies cannot be used by itself as a marker of lupus nephritis. Evidence of active consumption of serum complement proteins (e.g., low concentrations of C3 or C4; increased complement split products) may be seen in patients with active nephritis. Careful monitoring of the urine for signs of lupus activity (e.g., proteinuria, cellular casts, hematuria, pyuria) is an important part of the routine evaluation of patients with SLE. In addition, quantification of proteinuria (e.g., by a 24-hour urine collection) offers important information on the prognosis and response to therapy. For many patients, deciding how aggressively to treat lupus nephritis depends on its effects on the patient’s renal function. Therefore, it is important to note that the serum creatinine level or the creatinine clearance estimated from the creatinine concentration in a 24-hour collection may not truly reflect renal function and may overestimate the glomerular filtration rate in patients with lupus nephritis. If available more accurate determinations of glomerular filtration rate (e.g., inulin clearance) can be of value in assessing renal function in patients with lupus nephritis. Lastly, identifying and managing coexistant hypertension in lupus patients is cruicial to successful lupus management.
Therapy for lupus nephritis typically consists of corticosteroids in conjunction with immunosuppressive medications. Many patients are begun on high-dose steroids (e.g., 1 mg/kg prednisone) at the time of diagnosis of lupus nephritis. Depending on the other organ systems involved, this may be able to be tapered relatively rapidly. large boluses of corticosteroids (e.g., 1 g methylprednisolone on 3 successive days) are also used fairly commonly in an attempt to gain rapid control of disease activity. Based on the results of several prospective trials, immunosuppressive drugs have become the standard therapy for lupus nephritis. For many years, a typical regimen consisted of monthly boluses of cyclophosphamide (CYP; at a dose of approximately 750 mg/m2) for 6 months or longer, followed by additional boluses every 2 or 3 months, or an alternative immunosuppressant (e.g., azathioprine or mycophenolate) for a total of at least 2 years of treatment. Patients treated with shorter courses tended to have relapses of their disease; the ultimate length of treatment must be individualized based on the response as well as all of the manifestations of lupus active in the individual patient. Treatment with intermittent boluses of CYP became preferred to daily oral administration of the same medication because it was associated with fewer adverse effects, particularly hemorrhagic cystitis. Studies show that the “Euro-Lupus regimen” of six doses of CYP 500 mg given every 2 weeks (followed by azathioprine maintenance) yields equivalent outcomes compared to monthly pulse CYP 750 mg/m2 for 6 months, followed by quarterly infusions. Azathioprine and mycophenolate (mycophenolic acid and mycophenolate mofetil) have also been used successfully for the initial treatment of lupus nephritis. Because of its relative ease of use, mycophenolate has emerged as perhaps the most commonly used immunosuppressive agent. For patients with lupus glomerulonephritis with a rapidly progressive course, plasmapheresis is sometimes used in conjunction with cytotoxic therapy. Other drugs that have been used for the treatment of lupus nephritis include cyclosporine, the anti-CD20 monoclonal antibody rituximab and the T cell activation inhibitor abatacept. Rituximab and abatacept have been studied in randomized controlled trials with SLE and in lupus nephritis with negative results; however, this may relate in part to aspects of the study design. Observational data, particularly for rituximab, suggests that these therapies may have some efficacy.
—Neuropsychiatric: Neuropsychiatric manifestations of SLE are very common, with a prevalence of ~50%. Signs and symptoms can be quite varied (Table). It is often difficult to pinpoint SLE as the definite cause of many of these symptoms. Thus, an important part of the diagnosis involves excluding other potential causes, including infections (e.g., bacterial meningitis, viral encephalitis), medications (including psychotropic medications, high-dose corticosteroids, and NSAIDs), metabolic causes (e.g., the CNS effects of uremia), other medical conditions (e.g., hypertensive encephalopathy, seizure disorder unrelated to SLE, etc), and primary psychiatric disorders (e.g. depression). Confounding the diagnosis of neuropsychiatric SLE is the fact that there are no pathognomonic laboratory or imaging tests. Patients with CNS lupus may have abnormal CSF studies, including elevated CSF protein (including CSF concentrated or oligoclonal immunoglobulins) , elevated CSF cell counts, abnormal electroencephalograms, and various abnormalities on imaging studies (e.g., scattered high-intensity lesions in the white matter on T2 MRI). Although such testing can help exclude other causes and can be consistent with a diagnosis of CNS lupus, no test definitively establishes the diagnosis.
Treatment of neuropsychiatric SLE depends on the particular manifestations. Many patients, particularly those with severe involvement, receive corticosteroids or immunosuppressive agents. In addition, patients may benefit from therapies specific to their particular symptoms (e.g., antipsychotic medications for psychosis, antidepressants for depression, anticoagulants for thromboembolism and seizure controlling medications).
—Musculoskeletal: Musculoskeletal manifestations of SLE affect almost 90% of patients. Although most patients have arthralgia, fewer demonstrate an inflammatory arthritis (i.e. synovitis). The arthritis of SLE classically involves the small joints of the hands, wrists, and knees. In contrast to patients with rheumatoid arthritis, the arthritis is usually not associated with bony erosions observed on x-ray or fixed deformities. Some patients with SLE may develop changes in their joints that resemble those found in patients with RA (e.g., swan-neck deformity); however, unlike RA, such changes in SLE (known as Jaccoud’s arthropathy) are correctable or reducible on physical examination. Treatment of arthritis in patients with SLE often includes NSAIDs and the antimalarial drug hydroxychloroquine. Although patients often respond to corticosteroids, attempts should be made not to use them long term solely for arthritis. In patients with SLE with severe arthritis, treatment is comparable to that used for RA, and drugs such as MTX may be used. Patients with SLE with inflammatory myositis often require treatment with corticosteroids and other immunomodulatory drugs (e.g., azathioprine, methotrexate). Finally, osteonecrosis (e.g., hip, knee, shoulder) is seen among patients with SLE, particularly those treated with corticosteroids at high doses or for prolonged courses.
—Vascular: Vascular involvement is exceedingly common in SLE. Hypertension is among the most powerful predictors of renal survival and also overall patient survival in SLE. In addition, patients with SLE have excessive morbidity and mortality from atherosclerotic cardiovascular disease. Vasculitis with leukocytic infiltration and destruction of the involved vessel wall may rarely be seen in skin lesions and in other organ systems. More commonly, a bland vasculopathy is seen. Such lesions have alterations to the vessel wall and impingement of the vessel lumen without frank vasculitic changes (i.e. there is no disruption of the layers of the vessel). These changes are commonly seen in the CNS and other organ systems. Another factor that may predispose to thromboembolism is the presence of ACL antibodies.
| Table 1. Clinical Manifestations of Systemic Lupus Erythematosus |
| Constitutional symptoms – Fever, fatigue, malaise, anorexia, weight loss Mucocutaneous – Malar rash – Discoid rash – Photosensitivity – Oral/nasal ulcerations (typically painless at the onset) – Xerophthalmia (dry eyes) and/or xerostomia (dry mouth) (these sicca symptoms are consistent with Sjögren’s syndrome) – Alopecia (usually diffuse, in contrast to alopecia areata or male-pattern alopecia) – Lupus profundus (or lupus panniculitis; lupus mastitis when affecting the breast) Musculoskeletal – Arthritis (synovitis, commonly of PIP and knee joints) – Arthralgia – Fibromyalgia – Inflammatory myositis (with increased creatine kinase and proximal muscle weakness) – Osteonecrosis (particularly with chronic corticosteroid use) Renal/urologic – Glomerulonephritis (WHO classification: I, normal; II, mesangial; III, focal proliferative glomerulonephritis; IV, diffuse proliferative glomerulonephritis; V, membranous glomerulonephritis; VI, diffuse sclerosis) – Tubulointerstitial inflammation – Lupus cystitis (Note: Hemorrhagic cystitis is a potential complication of cyclophosphamide therapy) Hematologic – Lymphopenia (absolute lymphocyte count < 1500/mm3) – Leukopenia (WBC count < 4000/mm3) – Thrombocytopenia (platelet count < 100,000/mm3) – Hemolytic anemia (defined by positive Coombs test) – Lymphadenopathy (usually generalized) – Splenomegaly – Antiphospholipid antibody syndrome Neuropsychiatric – Headache (particularly refractory migraine-like headaches) – Seizures – Psychosis – Cerebral vascular accidents – Peripheral neuropathy – Cranial neuropathy – Transverse myelitis – Depression – Cognitive dysfunction Serosal – Pleuritis (exudative pleural effusion) – Pericarditis (exudative effusion; rarely associated with hemodynamic compromise) – Peritoneal inflammation (often presents with diffuse abdominal pain) Vascular – Raynaud’s phenomenon – Vasculitis – Vasculopathy (vessel wall abnormalities with minimal inflammation or damage) – Hypertension – Myocarditis – Endocarditis (e.g., Libman-Sacks lesions) – Thromboembolic events (particularly in LE patients with anticardiolipin antibodies or the lupus anticoagulant) Immunologic/laboratory tests – Antineutrophil antibody – False-positive nonspecific tests for syphilis (e.g., VDRL, RPR), anticardiolipin antibodies – Elevated serum concentrations of immune complexes – Evidence of complement consumption (e.g., decreased serum C3, C4 levels, or increased complement split products-C4b, C5a, sC5b-9) Miscellaneous – Pulmonary (pulmonary hemorrhage, pulmonary hypertension, interstitial lung disease, shrinking lung syndrome) – Ocular (cytoid bodies) – Gastrointestinal (lupoid hepatitis, pancreatitis, gastrointestinal vasculitis) |
Uncommon Findings: A number of distinct clinical syndromes are seen in patients with SLE.
—Drug-induced lupus: Patients treated with some medications may develop signs and symptoms of SLE. Drug-induced lupus is also associated with development of positive ANA, particularly with reactivity to histones. Indeed, the utility of a positive ANA cannot be determined in drug-induced lupus as a positive ANA is considered part of the diagnostic criteria for the disease. Also, many of the drugs implicated in drug-induced lupus cause a positive ANA in many more patients than those developing any clinical manifestations compatible with this syndrome. Common clinical features of drug-induced lupus typically include fever, arthritis, and serositis; lupus nephritis and CNS lupus are distinctly unusual in patients with drug-induced lupus. Agents strongly associated with drug-induced lupus include procainamide, hydralazine, and isoniazid. Others implicated include phenytoin and other seizure medications, quinidine, tetracyclines and TNF inhibitors. Regarding TNF inhibitors, a substantial minority of patients (approximately 30%) of RA patients receiving these agents will convert from a negative to a positive ANA. Uncommonly, anti-dsDNA and anticardiolipin antibodies are seen; but other autoantibodies are not. Drug induced lupus is uncommon with TNF inhibitor use (approximately 1/1000)
—Neonatal lupus: Neonatal lupus arises in newborns of mothers with anti-Ro (SS-A). Although some mothers have SLE or Sjögren’s syndrome, many are asymptomatic. When the antibodies cross the placenta (usually after week 16), they may bind cardiac tissue, resulting in heart block or myocarditis. Other manifestations may include rashes and thrombocytopenia, which may be transient and resolve as the maternal antibody disappears from the infant’s circulation.
Complications: Lupus is rarely complicated by thrombotic thrombocytopenic purpura (TTP) , Kikuchi’s disease, hemophagocytic syndrome or non-Hodgkins lymphoma.
Diagnostic Tests: Around 1948, the observation of the LE cell (a leukocyte that had engulfed another leukocyte) provided the basis for the ultimate definition of antinuclear antibodies (ANA) as the laboratory testing hallmark of SLE. The LE cell, which depended on the presence of very high titers of ANA, was relatively specific but insensitive for the diagnosis of SLE and is only of historic interest currently. It was replaced by immunofluorescent tests that looked specifically for the presence of antibodies capable of binding various nuclear constituents. Initially, ANA tests were performed on rodent tissue sections (e.g. rat or mouse liver or kidney). Of note, some nuclear antigens (e.g., Ro) are different in rodents, and some organelles (e.g., nucleoli, centromeres) are present in limited numbers in normal non-dividing tissue. Thus, in past years, there was a subset of “ANA negative” lupus patients who had clinical manifestations characteristic of SLE but were ANA negative. With the replacement of rodent tissues by the human HEp2 tumor cell as the standard substrate for ANA tests, the concept of ANA-negative lupus has largely disappeared. Although virtually all patients with SLE are positive for ANAs, the ANA test is very non-specific. Many patients with other connective tissue diseases and some healthy persons have ANAs, particularly at lower titers. In recent years, a variety of ELISA-based tests have replaced the fluorescent ANA test in many laboratories; the performance characteristics of these tests may differ from the HEp-2 ANA test.
Typically, fluorescent ANA test results are reported in terms of both titer and pattern. Higher titers are more consistent with, but not diagnostic of, SLE. Although normal value cut-offs are determined by local laboratories (and may differ), titers of 1:80 or higher are considered positive, whereas titers <1:80 are negative. Titers of the ANAs do not generally correlate with disease activity, and there is little value in repeating an ANA test in a patient known to be positive.
The patterns of immunofluorescence observed may correlate with different antigen reactivity (Table 2). A speckled pattern of immunofluorescence is the most common, but perhaps least specific. A speckled ANA is associated with various extractable nuclear antigens: Ro (SSA), La (SSB), Sm (anti-Smith), RNP, Scl-70, Jo-1, and many others. Anti-Ro and anti-La antibodies are also observed in patients with Sjögren syndrome (hence the designations SSA and SSB). Anti-Sm, when it done by radioimmunoassay, had been considered relatively specific for the diagnosis of SLE because it was seen infrequently in other diseases or in normal persons. However, by ELISA (as it is done mostly now) Sm test has become more sensitive but less specific. Along with anti-RNP antibodies, patients with SLE with anti-Sm may be more prone to develop interstitial lung disease. Anti-RNP antibodies have also been associated with MCTD . Anti-Scl-70 antibodies are associated with the diffuse form of systemic sclerosis.
A nucleolar pattern of the ANA is seen not only in SLE but also in inflammatory myositis and systemic sclerosis. A centromere pattern is associated with the limited form of systemic sclerosis (CREST syndrome). The homogeneous ANA is associated with antibodies to histones. Such antibodies had classically been associated with drug-induced lupus, but they are also commonly seen in SLE, and thus are of no value when differentiating idiopathic versus drug- induced lupus.
A rim pattern of immunofluorescence is associated with antibodies to native or double-stranded DNA. Anti-DNA antibodies, especially done by radioimmunoassay (the Farr assay) or on a particular organelle (the kinetoplast of Crithidiae lucillae) have been considered useful for the diagnosis of SLE because they were seen uncommonly in other diseases. In addition, patients with high titers of anti-DNA antibodies are more prone to develop proliferative lupus nephritis. The titer of anti-DNA antibodies may vary with the activity of disease, particularly lupus nephritis, and sequential determination of anti-DNA is sometimes used to follow the activity of SLE. However, more recently, with anti-dsDNA testing by ELISA, these associations may no longer be valid. Anti-dsDNA by ELISA tends to be more sensitive but less specific for SLE.
| Table 2. Correlations between ANA Pattern, Antigen Specificity and Clinical Associations | ||
| ANA Pattern | Antigen | Clinical Associations |
| Diffuse | Deoxyribonucleoprotein, Histones | Low titer = nonspecific; drug-induced lupus, SLE, Others |
| 50% of SLE (specific) | ||
| Peripheral | ds-DNA | |
| Speckled | U1-RNP | >90% of MCTD |
| Sm | 30% of SLE (specific) | |
| Ro (SSA) | Sjögren 60%, SCLE, Elder lupus, Neonatal lupus, ANA-negative lupus | |
| 50% of Sjögren, 15% SLE | ||
| La (SSB) | ||
| Scl-70 | 40% of PSS (diffuse disease) | |
| PM-Scl (PM-1) | Overlap scleroderma + myositis | |
| Jo-1 | Myositis, lung disease, arthritis | |
| Nucleolar | RNA polymerases | 40% of PSS |
| Centromere | Kinetochore | 75% CREST (limited disease) |
| Cytoplasmic (nonspecific) | Ro, ribosomal P, | Sjögren, SLE psychosis |
| Cardiolipin, | Thrombosis, fetal loss, ↓Platelets | |
| AMA, ASMA | Primary biliary cirrhosis, autoimmune hepatitis | |
| tRNA sythetases | Synthetase syndrome: Myositis, lung disease, arthritis | |
| Abbreviations: ANA, antinuclear antibody; ds-DNA, double-stranded DNA; LE, lupus erythematosus; SLE, systemic lupus erythematosus; SCLE, subacute cutaneous lupus; MCTD, mixed connective tissue disease; PSS, progressive systemic sclerosis; AMA, anti-mitochondrial antibody; ASMA, anti–smooth muscle antibody | ||
Diagnosis: The original 1982 classification criteria for SLE are shown in Table 3. A patient may be classified as having SLE if four or more of these 11 criteria are present at any time. Several relevant considerations affect the use of these criteria. First, they were developed to classify patients as having SLE rather than other autoimmune diseases such as scleroderma; therefore, some signs and symptoms that are very common not only among patients with SLE but also among patients with other autoimmune diseases (e.g., Raynaud phenomenon, alopecia) are not included. Second, the original criteria were developed in 1982, and some tests used then are no longer available widely (e.g., the LE cell preparation). Therefore, modifications have been suggested. For example, the SLICC group has proposed revised ACR Classification criteria that would allow 6 immunologic criteria: 1) ANA positive 2) anti-dsDNA above normal except for ELISA which would need to be twice the upper limit of normal 3) anti-Sm positive, 4) antiphospholipid antibodies (lupus anticoagulant, false positive test for syphilis, anticardiolipin antibody at least twice normal or ‘medium-high’ titer, 5) anti-beta2-glycoprotein-1 antibody and 6) direct Coombs test positivity in the absence of hemolytic anemia. In addition, the SLICC group added the clinical criteria of non-scarring alopecia, and separated hematologic criteria into 3 categories: 1) hemolytic anemia, 2) leukopenia or lymphopenia, and 3) thrombocytopenia.
| Table 3. American College of Rheumatology Criteria for the Classification of Systemic Lupus Erythematosus |
| 1. Malar rash 2. Discoid rash 3. Photosensitivity 4. Oral ulcers 5. Arthritis 6. Serositis 7. Renal disorder [persistent proteinuria (>0.5 g/day) or cellular casts] 8. Neurologic disorder (seizures or psychosis) 9. Hematologic disorder (hemolytic anemia, leukopenia, lymphopenia, or thrombocy- topenia) 10. Immunologic disorder (anti-DNA antibodies, anti-Sm antibodies, positive LE cell preparation) 11. Antinuclear antibody |
| A patient may be classified as having systemic lupus erythematosus if >4 of the 11 criteria are present at any time. |
Therapy: Treatment depends on the particular manifestation(s) for a given patient (see Table 1). Patients with arthritis or serositis frequently respond to NSAIDs. Antimalarials, particularly hydroxychloroquine, are effective for these same manifestations and are also used for SLE skin lesions. Despite concern about the adverse effects related to their use, corticosteroids are widely used for many manifestations of SLE. For skin disease, topical steroid preparations may suffice. For minor or moderate disease activity, doses of prednisone (<0.5 mg/kg) are often of great benefit. For severe manifestations (nephritis, pneumonitis, cerebritis, severe cytopenias), high-dose steroids (1 mg/kg prednisone) may be required. In all instances, because corticosteroids are associated with significant toxicity in SLE patients, attempts should be made to taper and discontinue steroids as rapidly as disease activity permits. Boluses of high doses (pulse dosing) of steroids (e.g., 250–1000 mg methylprednisolone daily for 3–4 days) have been used to gain rapid control of disease activity (e.g., cerebritis, nephritis, alveolar hemorrhage). For patients who require high doses of steroids for long periods of time, immunosuppressive drugs should be used as steroid-sparing agents. In addition to these immunomodulatory approaches, the optimal care of many patients with SLE also involves assiduous treatment of hypertension, treatment of clotting diatheses, and other general health measures.
BIBLIOGRAPHY
Hanly JG. Diagnosis and management of neuropsychiatric SLE. Nat Rev Rheumatol. 2014; 10: 338-47. PMID: 24514913
Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis 2014;73:1601-6. PMID: 24845390
Urowitz MB, Gladman DD, Ibanez D, et al. Evolution of disease burden ovr five years in a multicenter inception systemic lupus erythematosus cohort. Arthritis Care Res 2012;64:132-7. PMID: 21954226
Petri M, et al. The SLICC group revision of the ACR Classification criteria for SLE. Arthritis Rheum 2012; 64: 2677-86. PMID: 22553077
Houssiau FA, Vasconcelos C, D'Cruz D, et al. Early response to immunosuppressive therapy predicts good renal outcome in lupus nephritis: lessons from long-term followup of patients in the Euro-Lupus Nephritis Trial. Arthritis Rheum 2004; 50: 3934-40. PMID: 15593207
Boumpas DT, Austin HA 3rd, Vaughn EM, et al. Controlled trial of pulse methylprednisolone versus two regimens of pulse cyclophosphamide in severe lupus nephritis. Lancet 1992;340:741–745. PMID: 1356175
Karim MY, Alba P, Cuadrado MJ, et al. Mycophenolate mofetil for systemic lupus erythematosus refractory to other immunosuppressive agents. Rheumatology 2002;41:876–882. PMID: 12154204
Patel P, Werth V. Cutaneous lupus erythematosus: a review. Dermatol Clin 2002;20:373–385. PMID: 12170873
Wetter DA, Davis MD. Lupus-like syndrome attributable to anti-tumor necrosis factor alpha therapy in 14 patients during an 8-year period at Mayo Clinic. Mayo Clin Proc. 2009; 84): 979-84. PMID: 19880688